Computational Chemistry Services
Developing in silico solutions in the areas of de novo molecule design, ADMET predictions, Virtual screening, and Reaction modeling
We develop and deploy solutions that complement traditional lab-based approaches with in silico solutions to accelerate and efficiently tackle the complexity, scale, and richness of modern applications of computational chemistry. We reduce Avoidable Experimental Expenditure (AEE) by combining the concepts of cheminformatics and computational chemistry with advances in AI/ML, Cloud, and DevOps to organize, analyze, compute, and extract complex relationships from R&D data.
Our Strengths
Multi-Disciplinary Teams
Teams with blended skills in science (Biology, Chemistry) and technology (AI/ML, Data, Apps, and Cloud) help us seamlessly collaborate with our client’s biopharma R&D teams and build enterprise-class vertically integrated in silico solutions
Building Blocks
State-of-the-art Techniques
We work with proprietary and open-source tools, platforms, and technologies to expand the application of in silico techniques to emerging therapies and modalities. We leverage the latest advances and industry-validated approaches
Computational Chemistry - Solution Areas and Building Blocks
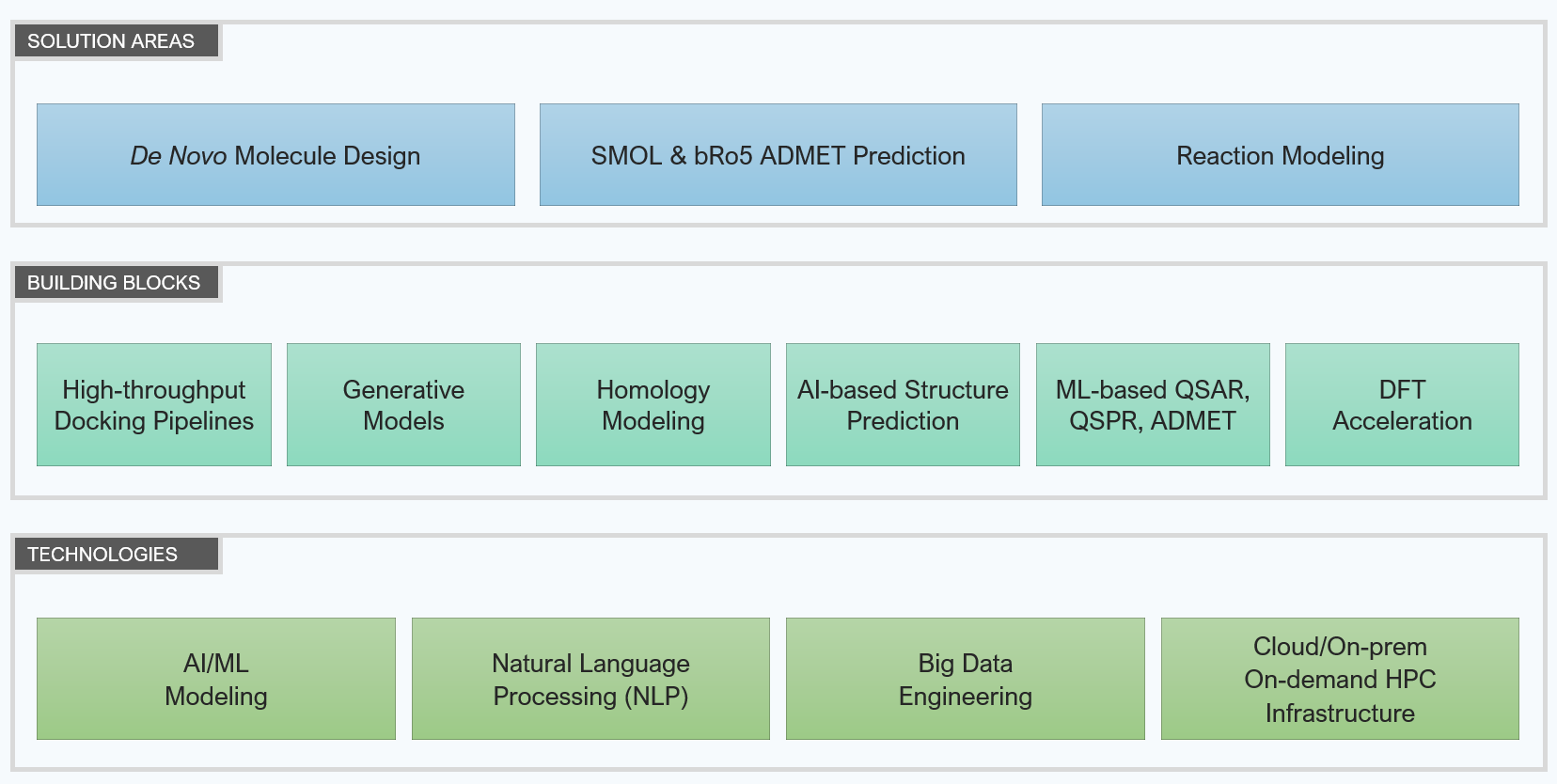
Solution areas
Solution area
De novo molecule design
We build solutions for de novo Molecule Design leveraging generative models to improve speed and optimize costs significantly. These solutions are cloud-based, repeatable, and optimized for synthesizability, drug-like characteristics, and biological activity.
Generative models
Navigate the vast ‘drug-like’ chemical space with generative models
- Models are optimized for synthesizability, drug-like characteristics, and ADMET properties.
High throughput molecular docking
Estimation of binding affinity of lead candidates through high-throughput molecular docking.
- Top docked complexes are further explored for potential interactions that reveal novel binding mechanisms through binding site analysis.
DFT acceleration
Leveraging computational pipelines for DFT acceleration
- Speeds up the estimation of DFT descriptors that help improve upon fingerprint-based ADMET predictions.
Structure Prediction
Structure prediction in cases where protein structure is not available through.
- Homology modeling.
- Deep-learning-based methods such as Alpha fold to predict the 3D structure of the protein
Solution area
ADMET prediction
ML-based QSAR and QSPR
ADMET prediction of New Chemical Entities (NCEs) with ML-based QSAR and QSPR
- Continuously retrain the models as more experimental data is available and periodically cluster the chemical spaces to investigate if models with high accuracy but narrow domain-of-applicability can be built.
- Fine-tune the models built on public datasets for enterprise data.
- Deploy the solutions on public cloud infrastructure or custom on-premise client environments or in hybrid models
DFT acceleration
Speeding up the estimation of energy, charge, bond length, etc., with DFT acceleration
- Enabling explainability of predictions using the electronic and geometric characteristics of the molecule such as EHOMO, ELUMO, Energy gap, IP, EA, electronegativity (𝜒), Chemical potential (μ).
Solution area
Reaction Modeling
Machine Learning Models
- Capture all the required descriptors (reactants, conditions, catalyst, base, ligand etc.) with machine learning models
NLP Pipelines
- Extract information from public datasets such as USPTO with NLP (Natural Language Processing) pipelines
Pre-trained models
- Pre-trained models curated from public datasets that are further fine-tuned on client provided proprietary data
DFT acceleration
- Estimate curated descriptors based on reaction-mechanism that help improve generalizability of models through DFT acceleration
Building Blocks
We have built building blocks to accelerate the development of new computational chemistry solutions
State-of-the-art AI/ML models
Homology modeling powered by RoseTTAfold and Deep learning based AlphaFold2 to construct tertiary and 3D structures of the target protein/antibody. DL/ML models to accurately predict the biological activity and ADMET properties of molecules spanning a diverse chemical space. DFT acceleration to study the electronic and geometric characteristics of the drug molecule
Generative model
Generative models that are powered by neural networks, reinforcement learning, and transformers/LSTM. These models are pre-trained on publicly available ‘drug-like’ datasets and generate new ’drug-like’ molecules. These models are optimized for multiple constraints such as synthesizability, toxicity, biological activity, etc.
High-throughput docking pipelines
Customizable and configurable high-throughput docking pipelines to screen for ligand molecules by predicting the protein-ligand binding affinity faster, and at scale. Powered by a Cloud-based Kubernetes environment to enable auto-scaling and schedulers for workload management
Scalable and on-demand computing infrastructure
Infrastructure-as-a-code driven solutions provide on-demand and auto-scalable computational resources with minimal technical effort. Compatible with any cloud provider supporting Kubernetes, including major vendors such as AWS, GCP, and Azure. Compatible with classic HPC schedulers such as SLURM and SGE for workload management
Download our case study on reaction modelling studies using QM/DFT methods for ligand optimization
Discover our offerings across the biopharma value chain
Our Solutions
Our Services
Offering services in computational sciences and technology to complement biopharma R&D