Understanding Drug Selectivity: A Computational Perspective
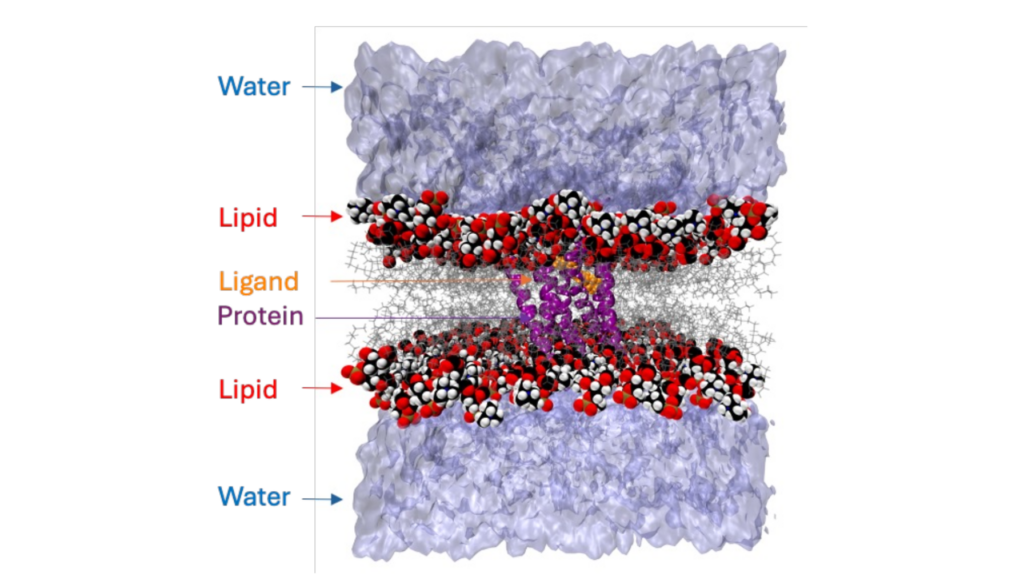
Importance of Drug Selectivity
Drug selectivity refers to the ability of a drug to preferentially interact with a specific target, such as a particular receptor, enzyme, or ion channel, over other potential targets in the body. High selectivity is often desired in drug design because it can minimize the likelihood of off-target effects, which can lead to adverse side effects. Biochemical and cellular assays provide valuable functional insights, however investigating the molecular origin of drug selectivity using experimental methods is resource intensive.
Bridging the Gap with Computational Studies
Scientists are increasingly using meticulously designed computational studies to complement experimental data. These studies offer valuable insights into ligand-receptor interactions, structural changes, and selectivity mechanisms. Advancements in computational infrastructure and the chemical accuracy of force fields have significantly improved the capabilities of molecular dynamics simulations. Today, researchers can explore the dynamic behavior of biomolecules with impressive detail and accuracy, achieving this within a reasonable timeframe. For example, the Anton 3 supercomputer, with its 512 nodes, can simulate a million atoms over a timescale exceeding 100 microseconds per day.
Case Study: Siponimod and S1PR1
Let’s focus on Siponimod, an FDA-approved drug used to treat relapsing forms of Multiple Sclerosis (MS). Siponimod targets sphingosine-1-phosphate receptor 1 (S1PR1), a member of the G Protein-Coupled Receptor (GPCR) family. Interestingly, Siponimod activates S1PR1 but not S1PR2, despite their structural similarity.
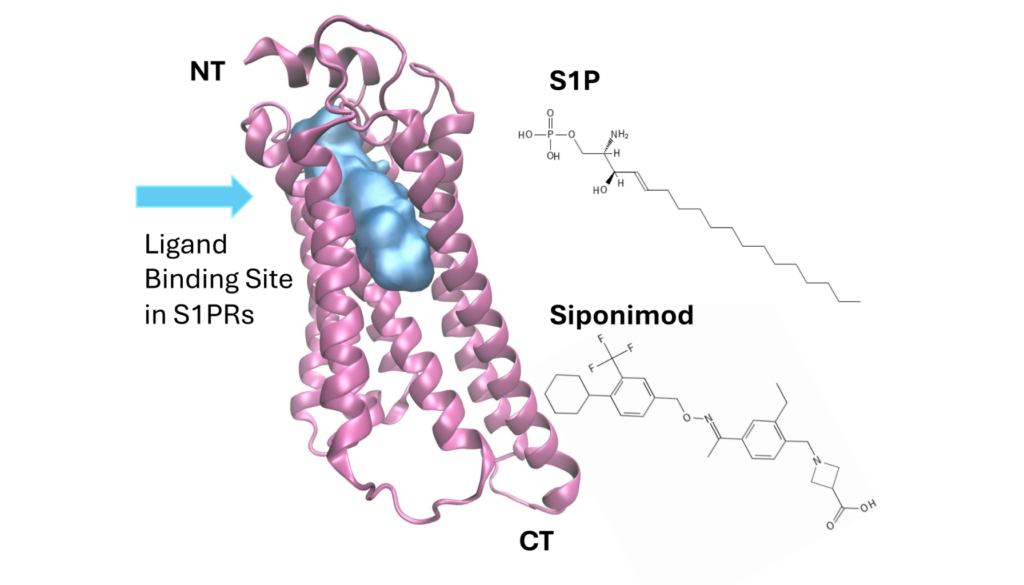
Contrasting Hypotheses
Previous hypotheses based on molecular docking suggested that Siponimod selectively binds to S1PR1 and S1PR5 but not to S1PR2 due to steric clashes. However, MD simulations revealed a different story. Siponimod can indeed bind to S1PR2 with an affinity comparable to S1PR1. So why the selectivity difference?
Unveiling the Mechanism
Comparing the dynamics of Siponimod-bound S1PR2 with S1P-bound S1PR2 provided insights. S1P activates transmission switches necessary for downstream biological activity in S1PR2 but Siponimod does not. Both S1P and Siponimod activate these switches in S1PR1. Siponimod, known to act on S1PR1 as a functional antagonist, could activate the transmission switches in S1PR1 in a similar manner as S1P does. But it fails to activate the transmission switches in S1PR2, thus S1PR2 fails to show any functional activity upon Siponimod binding.
Leveraging Residues for Selectivity
By identifying crucial residues in S1PR2, we can optimize molecules to selectively bind to S1PR1 over S1PR2. In silico approaches like MD simulations empower us to understand ligand-induced structural changes and guide drug development.
Conclusion
In summary, computational studies enhance our understanding of drug selectivity. They reveal intricate details that guide drug design. As we continue exploring GPCRs and other membrane proteins, in silico approaches remain invaluable tools in our quest for effective medications.
For further exploration, check out our recent publication titled as: “Selective Activation of GPCRs: Molecular Dynamics Investigation of Siponimod’s Interaction with S1PR1 and S1PR2”. Our work shows that MD powered in silico studies can be used to model and predict selectivity of candidate molecules to GPCRs, the largest family of membrane receptors targeted by approved drugs.