Target elucidation: Key first steps for structure based in silico drug design

BioPharma wins come after long and hard fought battles. Every phase of a drug discovery battle poses its own challenges. Deep Science and Deep Tech innovations are continuously expanding the options available for addressing these challenges.
For example:
Advances in functional genomics assays, combined with progress in development of AI foundation models to efficiently ingest and learn from large datasets produced by high-throughput assays, are enhancing the chances of success in identifying the right targets and validating them.
Similarly, advances in generative AI (Transformers, Diffusion/score-based) models are accelerating the design of therapeutics, across modalities – small molecules, antibodies, RNA, peptides, proteins, advanced gene and cell therapies.
AI models are also addressing drug development concerns such as PK/PD, crystal structure prediction, reaction modeling, and biosynthesis while new approach methodologies (NAM) such as organoids are accelerating safety studies.
This description may already seem dense, cutting across many disciplines, but this is still simplistic.
There’s a lot more that goes into preparing and executing these steps, even in early R&D. For example, before one can embark on a drug design exercise post target identification, it’s necessary to elucidate the target in several ways no matter which method of drug design is chosen. For a protein target, questions like the following need to be answered.
- What’s the biological context in which the target is located?
- Are there post-translational modifications and mutations to factor in?
- Does the target oligomerize? Should we target a specific oligomeric state?
- How many different conformations of the target exist? Which one should we target?
- Is there an activity of the target that we need to enhance or inhibit through the designed therapeutic? How is that activity triggered?
A number of de novo drug design requests we receive at Aganitha do not come with ready answers to such questions of importance. We hence pencil in a target elucidation phase at the beginning of our drug discovery engagements where we evaluate our targets for druggability and structural details using in silico approaches for various diseases. In this blog, we briefly illustrate the activities in the target elucidation phase for protein targets, with a few examples.
Selecting the oligomeric state to target:
Identifying optimal oligomeric state becomes crucial in cases where the protein target exists as a monomer as well as an oligomer. We consider factors like cellular accessibility and biological relevance to make an informed decision regarding the targeted oligomeric state.
Example 1 (Based on biological relevance):
Biological information available for the target and its homologs revealed that homodimer is the biologically active state. Through our druggabiltiy analysis of the modeled dimer, we discovered a binding pocket at the interface of this dimer.
Example 2 (Based on cellular localization):
One of our targets exhibited ATP-dependent dimerization. When present at the cell membrane, ATP bound protein is a dimer associated with other proteins as well. The same protein when bound to ADP is a monomer and not associated with other proteins. Based on cellular localization, the dimeric state was more complex while the monomeric form was relatively free and easy to target during cell division.
Selecting the conformation to target:
Protein function is highly dependent on the protein’s conformational landscape. We utilize in silico tools and molecular dynamics (MD) simulations to generate the repertoire of conformations for our target, with associated probabilities.
Example 1:
In Figure 2, we obtained different binding pocket conformations of target protein using computational methods. Conformations with high probabilities of occurrence were used in downstream drug design pipelines.
Example 2:
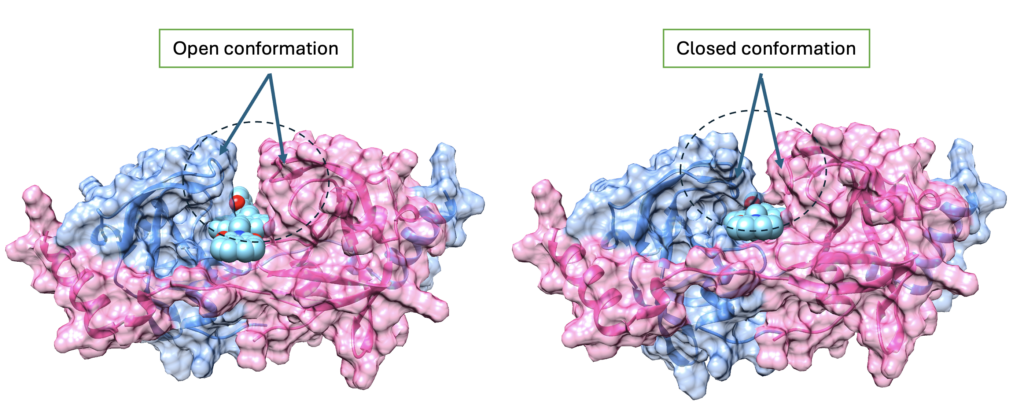
For another protein (Figure 3), closed and open conformations were observed with terminal loops acting as the hinges of a door. Closed conformation which can stabilize a molecule inside the binding pocket was used downstream.

Tackling a target with no a priori structural information:
SBDD is challenging when the structural information for target protein or any of the homologues is missing.
Example:
One of our targets is a transmembrane protein with no reliable structural information or known homologs. On top of that, no cognate ligands are known that bind the transmembrane protein. Conformational ensembles of the target protein in presence of a lipid membrane were generated to identify representative structures with potential binding sites. Binding sites will further be validated using AI/ML tools used to identify cryptic pockets.
We step into SBDD and Fragment Based Drug Discovery(FBDD) once we have a good structure with a desired conformation and oligomeric state. Surely, the de novo drug discovery process has its own set of challenges which we will bring about to your attention in upcoming blogs. Until then, stay tuned and explore our services and solutions at aganitha.ai.
P.S. (Aug 23, 2024):
The point that we made in this blog about the challenges of target structure elucidation even in the post AlphaFold era has once again been highlighted in a recent publication by Herrington, et al.
Similar to our experience, the authors found that high-confidence AF2 models often require further refinement, akin to many PDB structures, to address issues such as undesired side chain placement, occluded protein pockets, etc. They also emphasize the potential of MD simulations and induced-fit docking protocols as promising solutions.