De Novo PROTAC Design with Gen AI and FBDD
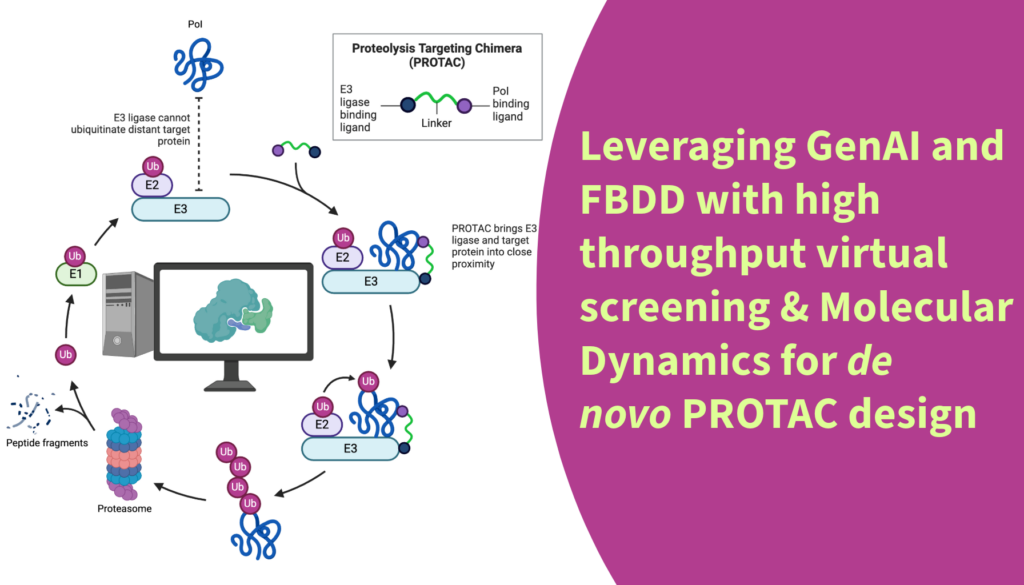
A PROTAC (PROteolysis TArgeting Chimera) is a small molecule that facilitates the degradation of disease-causing proteins by recruiting an E3 ubiquitin ligase to the target protein, leading to its ubiquitination and subsequent degradation by the proteasome. PROTAC technology effectively addresses undruggable targets and overcomes drug resistance. Its catalytic mechanism allows for high potency at low concentrations, potentially reducing toxicity and enhancing therapeutic efficacy.
Why designing PROTACs is a whole different ball game.
Designing effective PROTACs poses several challenges, starting with the selection of the right target protein and E3 ligase. Not all proteins are amenable to degradation via PROTACs, and the limited options for E3 ligases further constrain the design. Achieving stable ternary complex formation between the PROTAC, target protein, and E3 ligase is critical for success, requiring careful optimization of binding interactions, spatial arrangement, and cooperation. Additionally, PROTACs’ large molecular size and poor drug-like properties pose difficulties for oral bioavailability, cell permeability, and tissue penetration, complicating the balance between potency and pharmacokinetics. Ensuring selective degradation of target proteins while avoiding off-target effects and minimizing E3 ligase disruption—particularly the “hook effect” at high doses—also remains a significant challenge.
In silico methods show great promise for designing de novo PROTACs
Computational methods have the potential to greatly accelerate PROTAC design by leveraging high-quality experimental data. Screening of PROTAC libraries, traditionally a time-consuming task, can be streamlined through deep learning models that predict degradability of the ternary complex, helping researchers identify PROTACs with the highest potential for effective degradation.
Furthermore, Molecular Dynamics (MD) simulations provide insights into the dynamic behavior of the ternary complex (PROTAC, target protein, and E3 ligase), helping to optimize the spatial arrangement and stability of these complexes, which is critical for efficient degradation.
Additionally, computational approaches allow for de novo design of the ligand for the target protein and the selection of appropriate linker for an appropriate E3 ligase binder.
Overall, computational tools enable a faster, more efficient, and more accurate design process for PROTACs, reducing the need for labor-intensive experimental screening.
Where do we come in?
We, at Aganitha, have developed a solution to screen or design de novo PROTACs, leveraging deep learning with high throughput computational chemistry. We identify synthesizable fragments for each component—E3 ligase binder, protein of interest (POI) binder, and linker—ensuring they are compatible and capable of forming PROTACs with high predicted degradability. PROTACs with promising bioavailability are shortlisted, and our automated pipeline identifies the stable conformations of their ternary complexes. The dynamics of these stable complexes are then analyzed in detail to select the top candidates for further development.
We can assist you in designing PROTACs tailored to your specific needs, accelerating your drug discovery process. Interested in learning more? Contact us today. To learn more, visit about our PROTAC solution page. Stay tuned for an upcoming case study on designing PROTACs for specific disease targets.